ちょっとした機能
Atomsの操作関連
fmaxを返す
Imageのエネルギーのリストを返す
Imagesをコピーする
ImagesにCalculatorを取り付ける
ImagesにConstraintsをかける
Notebook上にエネルギーダイアグラムを作成する
NEBや振動数のエネルギーダイアグラムの可視化に使える.
from grrmpy.functions import draw_graph
draw_graph(images)
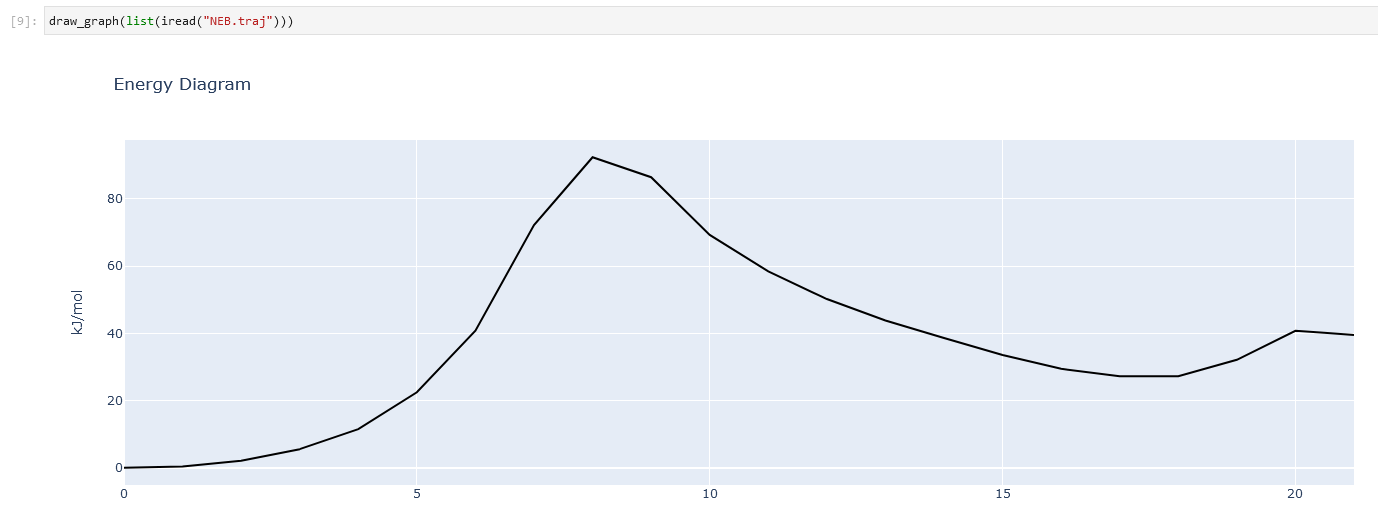
- grrmpy.functions.draw_graph(images, highlight=None, annotation=[], unit='kJ/mol', title='Energy Diagram', xaxis_title='', yaxis_title=None, calc_func=<function pfp_calculator>)[ソース]
エネルギーダイアグラムのグラフを作成する
Parameters:
- images: list of Atoms
- Atomsオブジェクトのリスト
- highlight: list of int
指定したindex番号のプロットのみ赤色になる
- annotation:list
- アノテーションを入れたい場合に設定[((x座標,y座標),"テキスト",{その他}),]のタプルのリストで与えるy座標もなくても良い,[x座標,テキスト]で与える{その他}はなくても良い.(2要素で与える)その他には例えば"showarrow":True 矢印で示す"arrowsize":2 矢印の大きさ"arrowhead":3 矢印の種類"ax:0" 矢印の向き"ay":-50 矢印の向きなど
- unit: string
- 'eV', 'kJ/mol', 'Hartree', 'kcal/mol'のいずれか
- xaxis_title: str
- x軸のタイトル
- yaxis_title: string
- Noneの場合,Energy({unit})
ユニークなCONNECTIONを返す
from grrmpy.io import read_connections
from grrmpy.functions import get_unique_connections
original_connections = read_connections("XXX_TS_list.log") #CONNECTIONを読み取る
connections,idx = get_unique_connections(original_connections)
# 元々どのindex番号なのかidxに入る
- grrmpy.functions.get_unique_connections(connections)[ソース]
ユニークなCONNECTIONSを返す
同じEQを繋ぐconneciotns,同じ組み合わせのconnections,'??'などを含むconnectionsを削除し 新しいconnectionsのリストを作成する.
戻り値は2要素のタプルで1要素目がユニークなconnections, 2要素目がそれが元々どのindex番号のconnectionだったのかを返す.
Parameters:
- connections: list of lists of int
- [[0,1],[1,0],[2,5],["??",2],[3,4]]のようなリストgrrmpy.io.read_connecitonsで取得できる
- 戻り値
ユニークなconnections, 元々のindex番号
- 戻り値の型
tuple
NEB関係
TSのindex番号を返す(&バリアレスの判定)
- grrmpy.neb.functions.get_imax(images, threshold=5, calc_func=<function pfp_calculator>) int[ソース]
TSのindex番号を返す
バリアレス(極大値・極小値: 0個)の場合Noneを返す.山が1つ(極大値: 1個,極小値: 0個)かつ,活性化障壁がthreshold[kJ/mol]以下であればバリアレスと考えNoneを返す.(正反応,逆反応のとちらかがthreshold[kJ/mol]以下の場合)Parameters:
- images: list of Atoms
- NEBイメージ
- threshold: float
- 活性化障壁がthreshold(kJ/mol)以下の場合はバリアレスと判定する
- calc_func: function object
- imagesにcalcuatorが設定されていない場合に必要
- 戻り値
TSのindex番号,バリアレスの場合None.
- 戻り値の型
int or None
活性化エネルギーを算出する
- grrmpy.neb.functions.get_ea(images, reverse=None, calc_func=<function pfp_calculator>, unit='kJ/mol')[ソース]
活性化エネルギーを算出する
Parameters:
- images: list of Atoms
- Atomsのリスト(NEBイメージ)
- reverse: bool
- Trueの場合,逆反応のEaを返す. Noneの場合,どちらか高い方を返す.
- calc_func: function object
- calculatorを返す関数
- unit: str
- 戻り値の単位(kJ/mol, eV, Hartreeのいずれか)
- 戻り値
Ea値
- 戻り値の型
float
NEBイメージの中の極小点のindex番号を返す
最適なimage数を算出する
- grrmpy.neb.functions.get_appropriate_nimages(ini_atoms, fin_atoms, dist=0.35, nmax=24, nmin=8, mic=None)[ソース]
最適なNEBのイメージ数を算出する.
最も移動距離の大きい原子が,d(Å)ずつ移動する時のイメージ数を返す. maxとminで最大,最小のイメージ数の制限を与えることができる.
Parameters:
- ini_atoms: Atoms
- Atoms
- fin_atoms: Atoms
- Atoms
- dist: float
- d(Å)毎にイメージ切る
- nmax: int
- 最大のimage数
- nmin:
- 最小のimage数.
- mic: bool
- 最小イメージ規則を適用するか.Noneの場合与えたAtomsが周期境界条件である時,自動でTureにする.
ジオメトリ関係
- grrmpy.functions.minimize_rotation_and_translation_for_specified_indices_only(target, atoms, indices=None)[ソース]
atomsの原子の位置をtargetの位置と近くなるように(最小二乗法)配置する.
ase.build.rotate.minimize_rotation_and_translationでindexを指定できるようにした関数 indices=Noneの時はASEのminimize_rotation_and_translationと同じで全ての原子を動かす.
結合状態を返す
分子を抽出する
- grrmpy.functions.connected_components(atoms, indices=None, mult=1.0, **kwargs)[ソース]
atoms中に存在する分子をindex番号毎にまとめたジェネレーターを返す
Exampleを参照
Parameters:
- atoms: Atoms
対象のAtoms
- indices: list of int
特定の原子のみcomponentを調べる時に指定する. Noneの場合,全ての原子を対象とする.
- mult: float
大きい程,離れていても結合していると判定される.
- kwargs:
- 元素毎で解離の共有結合半径を指定できるex) H=0.5 で水素の共有結合半径を0.5Åに変更できる.
サンプル
例えばatomsの[5,6,7,8,9]がメタン分子,[10,11]がNO分子だった場合, 分子ごとに分離することができる
>>> gen = connected_components(atoms, indices=[5,6,7,8,9,10,11]) >>> for i in gen: >>> print(i) >>> #--> {5,6,7,8,9} >>> #--> {10,11}
2構造の差分を算出する
リスト操作
リストをずらして取得する
l = [0,1,2,3,4,5,6,7,8,9]
list(functions(l,2,1))
# [(0, 1), (1, 2), (2, 3), (3, 4), (4, 5), (5, 6), (6, 7), (7, 8), (8, 9)]
list(functions(l,3,1))
# [(0, 1, 2),
# (1, 2, 3),
# (2, 3, 4),
# (3, 4, 5),
# (4, 5, 6),
# (5, 6, 7),
# (6, 7, 8),
# (7, 8, 9)]
list(functions(l,3,2))
# [(0, 1, 2), (2, 3, 4), (4, 5, 6), (6, 7, 8), (9,)]
list(functions(l,3,2,strict=True))
# [(0, 1, 2), (2, 3, 4), (4, 5, 6), (6, 7, 8)]